
Лаборатория структуры и функций генов человека
Разработка подходов к диагностике и генной терапии онкологических заболеваний (Т. В. Виноградова, Е. П. Копанцев, И. П. Чернов)
Большое внимание в последние годы уделяется проблемам создания лекарств нового поколения. В рамках федеральной целевой программы «Исследования и разработки по приоритетным направлениям развития научно-технологического комплекса России на 2007–2012 годы» с участием лаборатории осуществляется крупный комплексный проект «Разработка и выпуск опытных партий новых эффективных направленно-модифицированных терапевтических и диагностических средств постгеномной генерации для использования в онкологической практике». Целью этого проекта является создание и выпуск в клинику первых генно-терапевтических препаратов для лечения рака легкого и пищевода. Используемые в проекте подходы базируются на фундаментальных исследованиях и методических разработках, выполненных в лаборатории. Проект объединяет крупнейшие научные центры страны: Институт молекулярной генетики РАН, Российский онкологический научный центр им. Н. Н. Блохина РАМН, Институт биоорганической химии им. академиков М. М. Шемякина и Ю. А. Овчинникова, Новосибирский государственный университет, Институт биологии гена РАН, Институт эпидемиологии и микробиологии им. Н. Ф. Гамалеи РАМН, а также биотехнологические компании ЗАО «Евроген» и ЗАО «Биннофарм». В рамках проекта созданы конструкции, позволяющие осуществлять специфическую экспрессию терапевтических генов в раковых клетках, а также системы доставки терапевтических генов в опухоли.
Таким образом, была создана основа для разработки и производства новых эффективных медицинских препаратов для онкологии: терапевтических средств на основе генно-терапевтического подхода, терапевтических средств на основе биологически активных белков и диагностических препаратов.
Рак и развитие (М. В. Зиновьева, Т. В. Виноградова, Г. С. Монастырская)
Одним из важнейших результатов, полученных в рамках этого проекта, является демонстрация того, что гены, которые включаются в эмбриогенезе при развитии легких и пищевода, выключаются при превращении нормальных клеток в раковые и наоборот. Тем самым подтверждается гипотеза о том, что возникновение и развитие раковых клеток можно сравнить с эмбриональным развитием или самовоспроизводством и дифференцировкой стволовых клеток тех или иных тканей. Гены, имеющие дифференциальную экспрессию и при раке и при развитии, возможно, являются ключевыми для опухолеобразования. Исследование таких генов позволяет осуществлять рациональный выбор мишеней терапевтического воздействия: наиболее перспективными мишенями являются системы, регулирующие эмбриональное развитие и нарушенные при канцерогенезе.
Модифицированные олигонуклеотиды (В. К. Потапов)
Главным направлением работы группы в последние 3 года было разработка методов получения и синтез модифицированных нуклеозидов с измененной структурой основания или сахаро-фосфатного остова. Наиболее перспективным направлением является разработка подходов к твердофазному синтезу морфолино-олигонуклеотидов — аналогов, несущих в своем составе вместо рибозы остаток морфолина. Аналоги сохраняют структуру ДНК, образуют прочные комплементарные комплесы с природными олигонуклеотидами и являются незаряженными молекулами, устойчивыми к действию нуклеаз.
Второе направление — синтез олигонуклеотидов, несущих в определенных местах спейсеры заданной длины с тиольной группой, которые используются группой проф. Влодовера из NCI (Frederic) для изучения комплексов с интегразами HIV—1 и ASV методами рентгеноструктурного анализа.
Микроокружение опухоли и экспрессия генов (Плешкан Виктор, Антонова Дина)
По современным представлениям опухоль представляет собой сложную структуру, состоящую из собственно опухолевых клеток и клеток опухолевого микроокружения. Микроокружение, или строма, представляет собой опухоль ассоциированные фибробласты (ОАФ), иммунные клетки, цитокины и другие элементы. Строма необходима для поддержания опухолевой прогрессии, защиты и пролиферации опухолевых клеток. Многообразное взаимодействие опухолевых и стромальных клеток обеспечивает эти процессы. Различные паттерны экспрессии генов характерны для опухолевых и стромальных клеток. Многие из таких дифференциально экспрессирующихся генов являются ключевыми для взаимодействия «опухоль-строма».
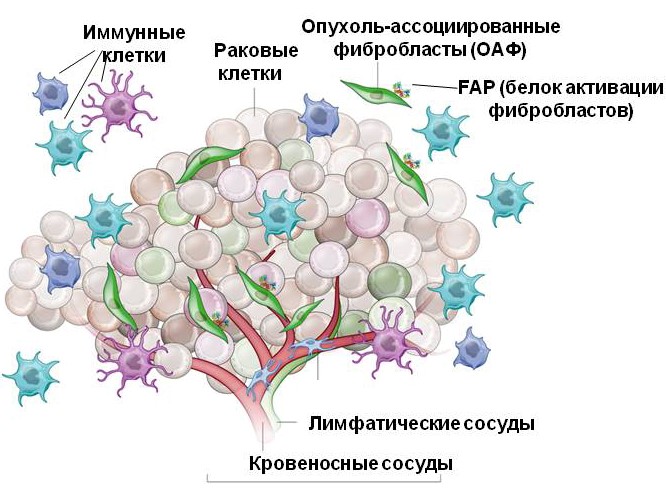
Опухолевое микроокружение (опухолевая строма) – это окружающие опухолевые клетки компоненты внеклеточного матрикса и неопухолевые клетки: эндотелиальные клетки, клетки иммунной системы, опухоль-ассоциированные фибробласты (ОАФ).
Нами исследуются ОАФ, их основные маркеры и способы воздействия на них, как на потенциальную мишень при противоопухолевой терапии. В Лаборатории были идентифицированы несколько генов, обладающих повышенной экспрессией в ОАФ. Мы исследуем промоторы этих генов на предмет тканеспецифичной экспрессии в клетках опухолевого микроокружения для использования в генной терапии. Также мы исследуем продукты генов опухолевого микроокружения и их роли при прогрессии опухоли и возможностях их использования для создания новых противоопухолевых подходов. Так, мы исследуем влияние фактора роста соединительной ткани CTGF на миграцию опухолевых клеток и его взаимосвязь c другими патологическими процессами при опухолевой прогрессии. В нашей группе проводятся эксперименты по выявлению потенциала опухоль-ассоциированных фибробластов, которые выступают в качестве мишени при противоопухолевой терапии. Мы готовы работать с новыми студентами, которые обладают возможностью и желанием усердно работать по данной тематике.
 Загрузка...
Загрузка...Научные проекты
Загрузка...Чернов Игорь Павлович
Москва, ул. Миклухо-Маклая, 16/10 — На карте
Cекретарь-референт
Левитан Татьяна Львовна
- Тел.: +7 (495) 330-65-29
- Факс: +7 (495) 330-65-38
- Эл. почта: levitanibh@mail.ru
Загрузка...