
Press-room / news / Science news /
Estimation of copy number variability from the high-throughput sequencing data
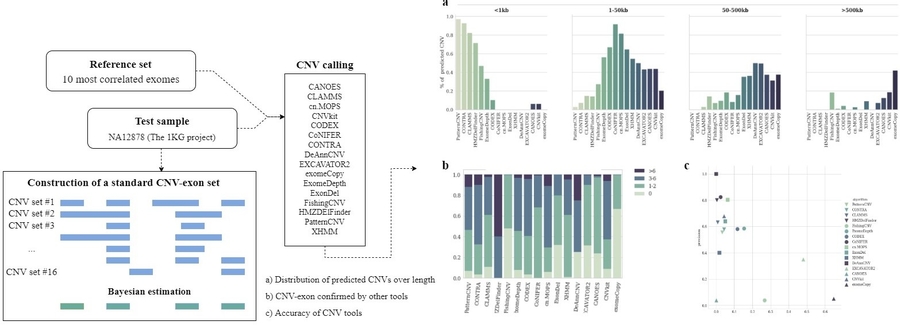
Traditionally, cytogenetic and molecular cytogenetic methods are used to detect chromosomal abnormalities. With the development of sequencing technologies, new approaches have become available to identify structural variations ranging from 50 bp. Researchers from the Laboratory of Molecular Oncology of Institute of Bioorganic Chemistry of the Russian Academy of Sciences and the Federal Research and Clinical Center of Physical-Chemical Medicine of Federal Medical Biological Agency developed an approach for construction of CNV validation set at the exon level and evaluated the efficiency of CNV calling tools designed for whole exome sequencing data.


Copy number variation (CNV) is one of the important sources of genomic variability, present in all mammalian species and involved in the processes of evolutionary adaptation, genomic disorders, and disease progression. The staff of the Laboratory of Molecular Oncology of the Institute of Bioorganic Chemistry of the Russian Academy of Sciences, together with colleagues from the Federal Research and Clinical Center of Physical-Chemical Medicine of Federal Medical Biological Agency published a number of papers ([1], [2]) devoted to the identification of CNV. Due to the wide variety of possible genomic variants, it is often necessary to use several approaches at once to detect them. The review describes in detail the main principles of modern methods, their advantages and limitations; among those presented there are karyotyping, chromosomal microarray analysis, analysis of sequencing data, optical mapping, etc.
Of particular interest is whole exome sequencing, which is widely used to identify single nucleotide variations and short insertions/deletions. However, the efficiency of using this technology to search for structural genomic alterations remains unclear. It has been shown that the CNV calling tools offered today are not equivalent. Each algorithm had a certain range of detected lengths and showed low concordance with other tools. Most tools are focused on detection of a limited number of CNVs one to seven exons long with a false-positive rate below 50%. EXCAVATOR2, exomeCopy, and FishingCNV focused on detection of a wide range of variations but showed low precision. In view of the different directions of algorithms, it is recommended to choose the most optimal one based on the study design and acceptable accuracy criteria.
march 14, 2022