
Пресс-центр / новости / Наука /
Исследования вариабельности числа копий фрагментов ДНК по данным высокопроизводительного секвенирования
Традиционно для детекции хромосомных аномалий применяют цитогенетические и молекулярно-цитогенетические методы. С развитием технологий секвенирования стали доступны новые подходы, позволяющие идентифицировать структурные вариации размером от 50 пн. Сотрудники Лаборатории молекулярной онкологии ИБХ РАН совместно с коллегами из ФГБУ ФНКЦ ФХМ ФМБА России разработали подход для формирования валидационного набора на уровне экзонов и оценили эффективность алгоритмов, предназначенных для данных полноэкзомного секвенирования.


Вариации по числу копий (CNV) - один из важных источников геномной изменчивости, представленный у всех видов млекопитающих и участвующий в процессах эволюционной адаптации, а также развитии заболеваний. Сотрудники Лаборатории молекулярной онкологии ИБХ РАН совместно с Коллегами из ФНКЦ Физико-Химической Медицины ФМБА опубликовали ряд работ ([1], [2]), посвященных вопросам идентификации CNV. Большое разнообразие возможных геномных вариантов зачастую требует применения сразу нескольких подходов для их обнаружения. В обзоре подробно описаны основные принципы современных методов, их преимущества и ограничения; среди представленных там - кариотипирование, хромосомный микроматричный анализ, анализ данных секвенирования второго и третьего поколений, оптическое картирование и др.
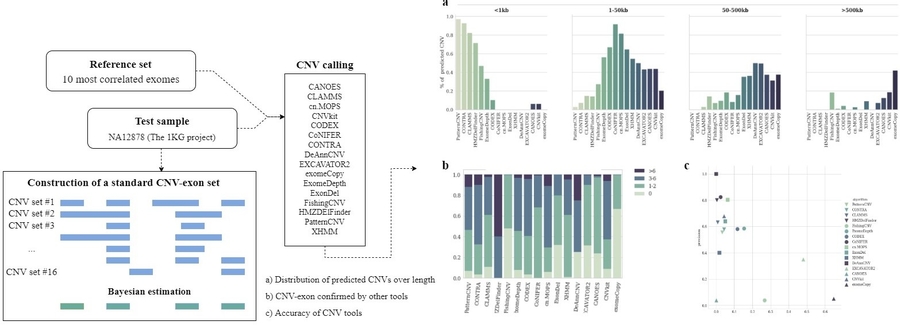
Особый интерес представляют данные полноэкзомного секвенирования, которое широко используется для поиска однонуклеотидных полиморфизмов и коротких инсерций/делеций. Однако эффективность применения данной технологии для поиска структурных перестроек остается неясной. Было показано, что предлагаемые сегодня инструменты неравнозначны - каждый из них имеет определенный диапазон детектируемых длин, а результаты предсказаний плохо согласуются между собой. Большинство алгоритмов ориентировано на поиск ограниченного числа CNV длиной от одного до семи экзонов, доля ложноположительных результатов не выше 50%. EXCAVATOR2, exomeCopy и FishingCNV способны идентифицировать широкий спектр вариаций, однако показывают низкую точность. Ввиду разной направленности алгоритмов выбор наиболее оптимального рекомендуется проводить исходя из дизайна исследования и допустимых критериев точности.
14 марта 2022 года